新加坡医疗器械四种审核路径满足不同企业注册需求
2022-07-08

新加坡政府致力于满足国内医疗保健需求,制定了一项长期计划,将医疗保健方面的 GDP 支出从目前的 4.6% 提高到 8%。到 2020 年,国民健康支出预计将增至每年 96 亿美元。新加坡在卓越医疗保健方面拥有强大的基础,提供强大的基础设施和全民健康覆盖,进一步增加了新加坡医疗器械市场的需求。
一、主管机构和核心法规
自2011年8月新加坡健康产品(医疗器械)法规开始实施,所有医疗器械如果想在新加坡销售,则必须经过新加坡卫生科学局(Health Sciences Authority),简称HSA,下属医疗器械部门认证。
HSA根据新加坡《健康产品法》(HPA,HEALTH PRODUCTS ACT 2007,2020修订版) 及其2010 年健康产品(医疗器械)HEALTH PRODUCTS ACT(CHAPTER 122D)条例对新加坡的医疗器械进行监管。在新加坡申请产品注册时,申请必须由同时作为许可证持有人或注册人的当地实体提交。
二、产品分类
在新加坡,医疗设备是指制造商打算单独或组合用于人类的任何仪器、装置、器具、机器、器具、植入物、体外试剂或校准器、软件、材料或其他类似或相关物品以诊断、预防、监测、治疗或减轻疾病或伤害为目的的生物。
HSA 严格遵循 GHTF 医疗器械分类指南,风险等级,从低到高分别为A、B、C 和 D 类不等。在新加坡,有四种不同的注册途径。所有医疗器械在新加坡供应之前都需要向HSA注册 ,但A 类低风险医疗器械除外,它们免于产品注册,或在HSA特别批准下豁免。另外,若已在某个参考国家/地区注册的 B、C 和 D 类设备有资格申请简化注册。未获得参考国家/地区批准的设备需经过完整注册流程。
三、认证模式
A 类无菌、B 类、C 类和 D 类医疗器械必须先提交给 HSA,然后才能在新加坡供应。HSA为企业提供了四条认证路径以满足不同企业需求:
1. 完整评估途径 (完整):
未获得任何参考监管机构事先批准的医疗器械的评估途径。
2. 简化的评估途径(简易):
已获得参考监管机构至少一项批准的医疗器械的评估途径(TGA – 澳大利亚、HC – 加拿大、MHLW – 日本、FDA – 美国、EC 证书 – 欧盟公告机构)
3. 即时/快速评估途径(加急) ——医疗器械的评估途径:
已获得参考监管机构的至少一项批准(D 类为两项)
在参考监管机构管辖下已上市至少 3 年
过去 3 年在全球范围内没有安全问题
没有事先被参考监管机构拒绝/撤回
4. 优先审查计划(完整优先):
优先审查计划为申请人提供了更快的注册和市场准入选项,以便通过全面评估途径提交给 HSA 的医疗器械获得更快的注册和市场准入。该优先审查仅适用于预期用途集中在以下五个医疗保健领域的医疗器械:癌症、糖尿病、眼科、心血管疾病和传染病。
具体各风险类别产品对应的审核路径:
1. MD/IVD A 类免于产品注册
2. MD/IVD B 类
即时:提交后立即注册
简易:100个工作日
全程:160个工作日
优先:120个工作日
3. MD/IVD C 类
即时:提交即刻注册(仅适用于C类独立医疗移动应用程序)
加急:120个工作日
简易:160个工作日
全程:220个工作日
优先:165个工作日
4. MD/IVD D 类
加急:180个工作日
简易:220个工作日
全程:310个工作日
优先:235个工作日
5. MD D 类 – 包含医药产品
简易:220个工作日
全程:310个工作日
注意事项
1. C 类设备排除在加急注册 (ECR) 之外:
髋关节、膝关节和肩关节置换术
非生物活性植入物。
2. 排除在加急注册 (EDR) 之外的 D 类设备:
有源植入设备
可植入设备连接循环系统或中枢神经系统
髋关节、膝关节和肩关节置换术(生物活性植入物)
将可注册药物纳入辅助角色的设备
HIV IVD 设备
血液/组织供体的兼容性
3. 参考国家批准要求
参考国家批准可以显著影响 B、C 和 D 类设备的注册途径。可接受的参考国家分别是澳大利亚 – 加拿大 – 欧盟 – 日本 – 美国。
4. 有效期和续订
只要维持年度上市费用并满足上市后的警惕性要求,注册就不会过期。
四、审核流程
新加坡允许将设备分组并命名为 Single、Family、System、Test kit 或 Group。产品注册档案必须按照东盟共同提交档案模板 (CSDT) 准备。在新加坡,申请必须通过医疗器械信息和通信系统 (MEDICS) 在线提交。在提交之前,公司必须申请 CRIS(客户注册和识别服务) 帐户并注册为 CRIS 管理员才能访问 MEDICS。
申请企业必须是本地实体公司,否则,需授权当地实体公司作为授权代表。指定的进口商和批发商必须拥有符合 GDPMDS 或 ISO 13485 的经销商许可证。GDPMDS 认证应从新加坡认可委员会认可的认证机构获得。A 类非无菌医疗器械需要简单的通知,经销商许可证仍需为当地实体公司。
具体注册流程如下: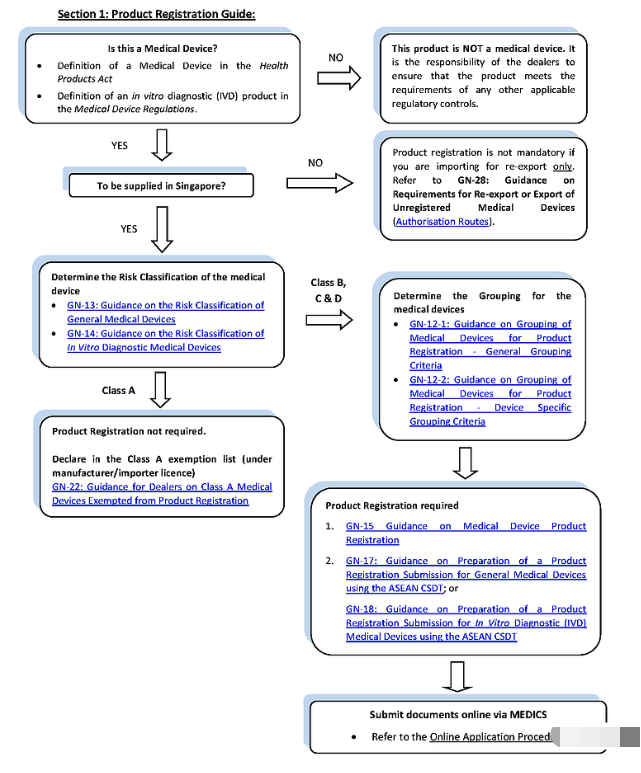
线上申请流程: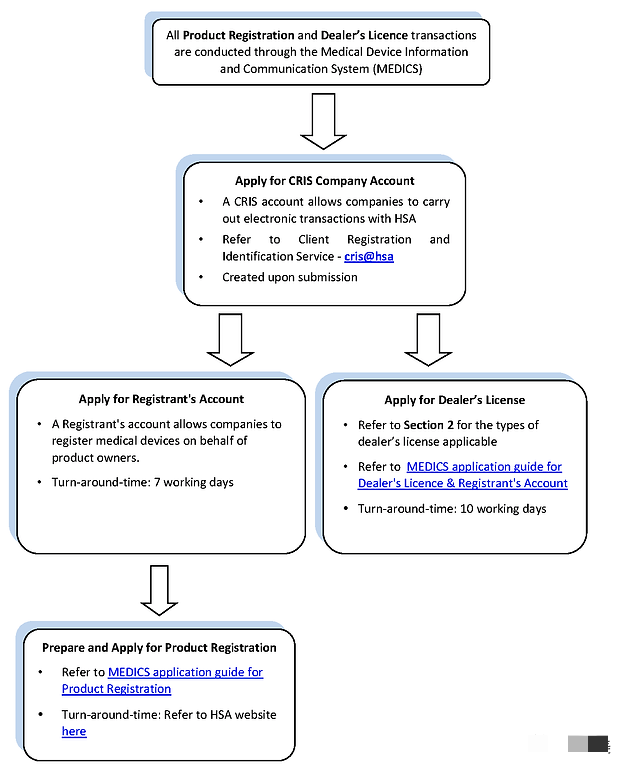
经销商许可证申请要求: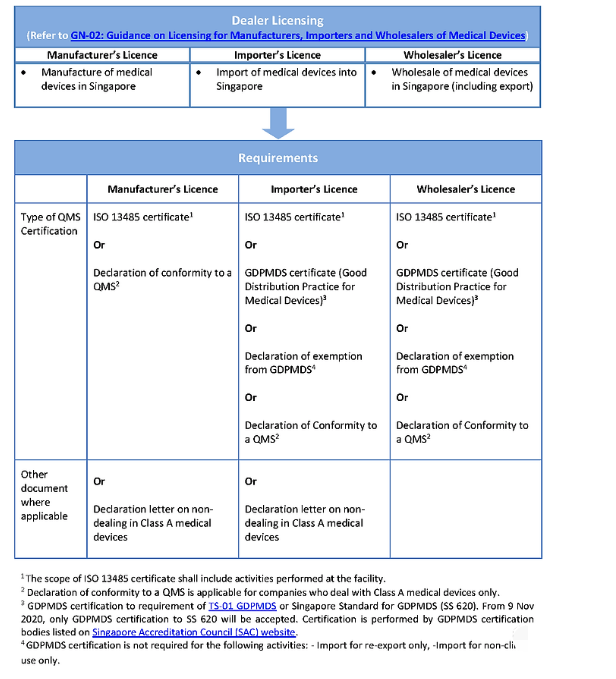
注意事项
1. 设备符合性评估
A 类设备不需要上市前注册,但必须由获得许可的进口商每年在 HSA 中列出。
HSA 提交档案基于东盟 CSDT(通用提交档案格式)。所需信息因设备分类和可用注册途径而异。除了欧盟技术文件要求的文件外,申请还需要一份基本原则符合性声明。
2. 质量体系合格评定
质量体系合格评定必须使用 ISO 13485 或 FDA 的审核报告(例如,企业检查报告)或日本 PMDA 进行记录。
3. 许可证持有人要求
当地代理人或注册人需要提交注册申请并持有许可证。此外,清关需要有执照的进口商。许可进口商需要 ISO 13485 和/或良好分销规范医疗器械 – 新加坡 (GDPMD-S) 许可证,并且必须在 HSA 注册。
4. 许可证转让
医疗器械注册申请必须由同时作为许可证持有人或注册人的当地实体提交。注册人是HSA 有关设备列表的主要联系人。《健康产品法》规定了与有效上市相关的注册人转让。指南在 GN-24,注册人变更指南中提供。
