医美产品出口韩国要求
2022-07-08
一、韩国主管机构及其法规
韩国卫生福利部(Ministry of Health and Welfare,MHW),简称卫生部,主要负责管食品、药品、化妆品和医疗器械的管理,是最主要的卫生保健部门。依照《医疗器械法》,韩国卫生福利部下属的食品药品安全部(MFDS)负责对医疗器械的监管工作,旧称KFDA(韩国食品和药品管理局)。
韩国对医疗器械根据产品风险等级不同分为4个等级: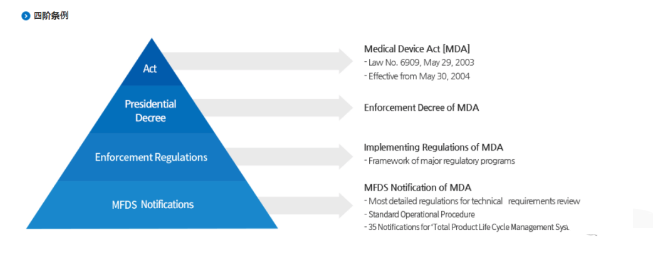
二、产品分类
根据 MFDS 第 2021-24 号通知,在韩国,设备根据其风险等级分为 I、II、III 和 IV 类。I 类设备对患者的风险很小,而 IV 类设备是高风险的复杂设备。
三、认证模式及审核流程
3.1 I 类和 II 类设备由国家医疗设备安全信息研究所 (NIDS) 的“医疗设备信息和技术援助中心”(MDITAC) 认证
产品分类:普通I类,Class I,监管路径:上市前通知。
韩国许可证持有人协助通过韩国海关清关产品。进口时必须出示上市前批准/通知许可证和KGMP证书,普通一类产品免于技术审查和KGMP认证。但I类无菌、一类测量,及其一类重复使用设备监管方式与II类设备相同。
普通I类医疗器械制造商可以在将注册信息上传到MFDS电子门户后完成产品注册。文件包括与医疗器械质量有关的文件。
准备材料:性能、安全等,预期用途、工作原理(MoA)、操作(功能)结构、原材料、使用说明、测试规范等信息。所有文件必须是韩文。
注册时间周期:大约2个月
3.2 III 类和 IV 类设备由 MFDS 批准。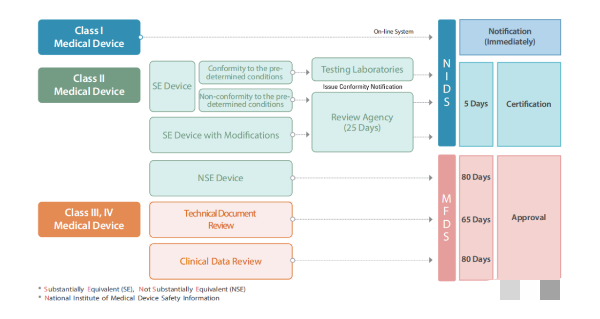
3.2.1 产品分类:II类器械,监管路径:上市前认证。
II类器械认证方式与美国FDA 510K类似,以比对产品方式证明产品安全及有效性。
材料准备:认证产品是在预期用途、工作原理 (MoA)、原材料、性能、测试规范(不适用于 IVD)、使用说明(不适用于体外诊断)等各方面与比与先前批准/认证/通知的医疗器(比对产品)等效的医疗设备。
3.2.2 改良器械(和对比产品有差异)
材料准备:预期用途、工作原理 (MoA)、原材料与先前批准/认证/通知的医疗器械等效,但在原材料(仅适用于 IVD)性能、测试规范(不适用于 IVD)、使用说明(不适用于 IVD)等不同或有所改进。
注册时间:II类常规-6个月,安全有效性审查——15个月
体系现场审核要求:II类以上产品制造商和韩国许可证持有人都必须遵守韩国KGMP质量体系要求。KGMP包括对国外制造设施的现场审核。由MFDS颁发的KGMP证书,有效期为3年。在产品注册之前需要KGMP认证。
3.3.3 III类和IV类,监管路径:上市前批准
对于II类、III类和IV类设备,MFDS将需要以下两种审查方案:
(1)一般技术文件审查:如果设备与合法销售的设备基本相同,则不需要“临床试验报告”。
(2)技术安全和效力审查(SER)。对于技术性SER,临床研究报告是必不可少的要求。如果“预期用途”、“工作原理(MoA)”和“原材料”等差异可能显着影响设备的安全性和有效性,则需要“临床试验报告”。
技术文件或安全有效性评审(SER)。临床数据必须提交给医疗服务部。MFDS可以接受从其他市场获得的临床数据。但所有文件必须是韩文。
另外,需向独立实验室(如韩国测试实验室)提交设备进行型式试验,或提交现有等效试验结果。如果您现有的(国外)测试报告符合MFDS测试标准,则不需要在韩国完成本地类型测试。
一般技术文件或SER技术文件由MFDS审查。
注册时间:III类定期–9个月,安全有效性审查——15个月
IV类常规——9个月,IV级,安全有效性审查——15个月
四、注意事项
1. 韩国证书持证人(Korea license holder) 简称:KLH
除了产品注册,您需要指定在韩国注册的证书持有人,该持有人必须获得KGMP认证。
2. 韩国生产质量管理规范(KGMP)
除了产品注册外,体系必须满足韩国国家KGMP认证。
KGMP的内容与ISO 13485基本相同。如果企业有符合ISO 13485体系,只需对现有体系稍加修改和/或补充符合韩国法规的要求的体系内容,即可满足现场KGMP要求。
