澳大利亚医疗器械认证简介
2022-07-081主管机构和核心法规澳大利亚的医疗器械,普通医疗器械和IVD产品需在澳大利亚药品注册库(ARTG)中列名后,所有在澳大利亚销售的医疗器械必须在澳大利亚医疗用品管理局(TherapeuticGoodsAdministration,简称TGA注册)TGA是发证机构也是医疗用品管理局,对此业内把医疗器械产品进行澳大利亚TGA注册的过程称为TGA认证。

1 主管机构和核心法规
澳大利亚的医疗器械,在澳大利亚药品管理局 (TGA) 监管下,受1989年《治疗品法案》(Therapeutic Goods Act 1989)和2002年《治疗品(医疗器械)条例》(Therapeutic Goods (Medical Devices) Regulations 2002 )的管制。
普通医疗器械和 IVD产品需在澳大利亚药品注册库(ARTG) 中列名后,才能在澳大利亚上市。
根据澳大利亚《医疗用品法》及《医疗用品(医疗器械)条例》等法规要求,所有在澳大利亚销售的医疗器械必须在澳大利亚医疗用品管理局(Therapeutic Goods Administration,简称TGA注册)TGA是发证机构也是医疗用品管理局,它是澳大利亚的医疗用品(包括药物,医疗器械、基因科技和血液制品)的监督机构。对此业内把医疗器械产品进行澳大利亚TGA注册的过程称为TGA认证。
2 产品分类
上述治疗品法案及条例41BD部分阐述了澳大利亚对医疗器械的分类,类似于澳大利亚的分类规则和标准。医疗器械和体外诊断器械有着不同的分类。医疗器械根据对人体造成可能风险的高低,由低到高,分为四类,即I类、IIa类、IIb类和III类。IVD产品分为1类IVD、2类IVD、3类IVD和4类IVD。
3 认证模式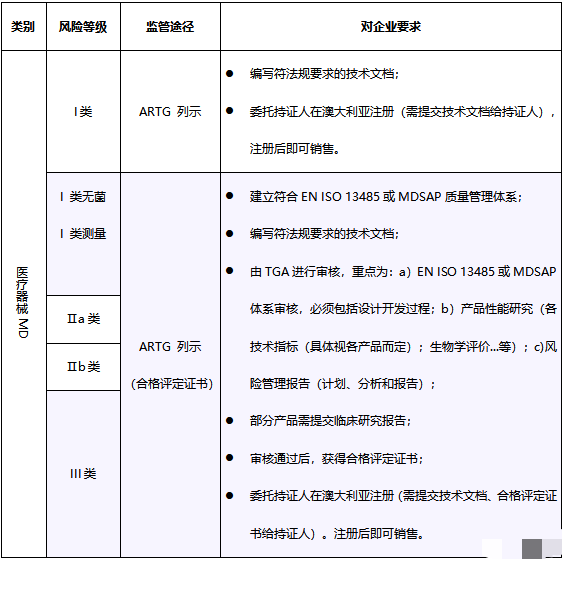
4 审核流程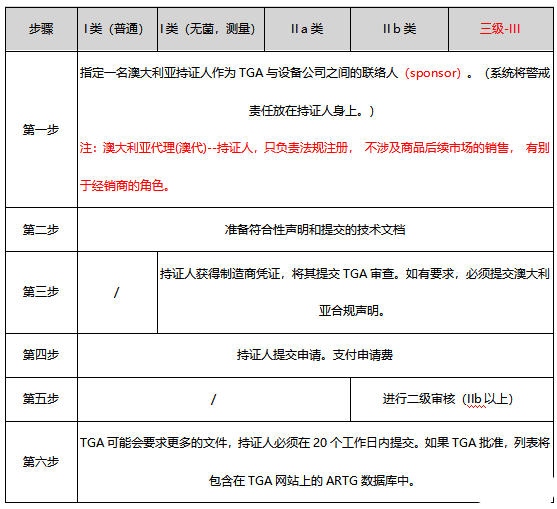
简化程序—— 一些海外证据(如MDSAP、美国FDA PMA/510K、加拿大卫生部HC MDL、日本MHLW等颁发的证书日本PMDA或RCB的PMA), 欧盟公告机构颁发ISO 13485:2016 证书,可用于支持符合性评估以简化TGA审核或注册流程。
高风险医疗器械需要TGA符合性评估,而CE认证不是合规性的可接受证据。
