马来西亚医疗器械认证不再需要FSC
2022-07-08

作为快速增长的医疗器械市场之一,近年来,马来西亚努力改善民众获得医疗服务的机会,马来西亚投资发展局(MIDA)将医疗器械行业确定为第十一个马来西亚计划下的高增长行业,于 2016-2020 年生效。
目前,马来西亚国内医疗器械行业集中于医用手套和其它一次性产品,而其它医疗器械严重依赖外国进口,从而使其成为大多数医疗器械制造商的竞争市场。
1 主管机构和核心法规
根据于 2013 年 7 月 1 日生效的 2012 年医疗器械法的规定,马来西亚的医疗器械产品注册由马来西亚卫生部 (MoHM)下属的医疗器械管理局(MDA) 监督。根据该法案,医疗器械在进口和投放市场之前需要在 MDA 注册。
在马来西亚,医疗器械是制造商打算单独或组合用于人类的任何仪器、装置、器具、机器、植入物、体外试剂或校准器、软件、材料或其他类似或相关物品以诊断、预防、监测、治疗或减轻疾病或伤害为目的的器械。
2 产品分类
根据 2012 年医疗器械法规,MDA创建了一个与东盟 MDD密切相关的分类系统。可能影响医疗器械分类的风险级别取决于预期用途、设计、制造和使用过程中应用的风险管理技术的有效性、预期用户、操作模式和使用的技术。
医疗器械产品按风险从低到高分为以下四类:
A级
B类
C类
D类
IVD 与其他医疗器械分开分类,风险取决于预期用途、预期用户的专业知识、诊断结果信息的重要性以及测试结果的影响。风险由低到高分别为:
A级
B类
C类
D类
例如,妊娠试验是 B 类,而 HIV 血液检测是 D 类。
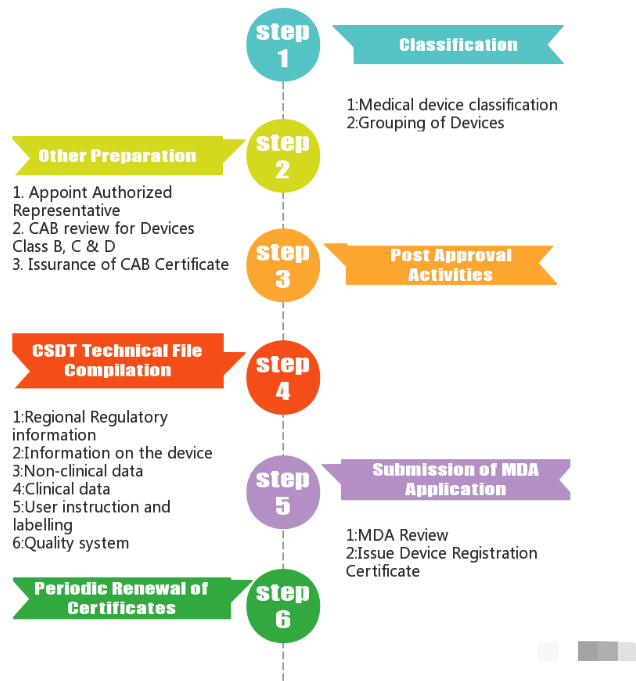
3 认证模式
非马来西亚本地医疗器械制造商需要在其境内有当地企业作为其代表,被称为医疗器械授权代表AR。他将成为制造商和医疗器械管理局(MDA)之间的联络人,以进行注册和提交申请。授权代表AR必须拥有营业执照和医疗器械良好分销规范 (GDPMD) 证书。
1. A类产品
由当地授权代表AR向MDA申请A类产品的注册,无需MDA授权的合格评定机构CAB批准。但是申请人必须提交制造商的ISO13485证书、测试报告以及标签等文件。
2. B、C、D类产品
当地授权代表AR提交B、C、D级产品的技术报告。由CAB进行技术文件审查。
得到参考国(如澳大利亚、加拿大、欧盟、日本、美国)批准和销售的医疗器械将通过简化程序审核,审核时,需向CAB提交ISO证书、CE证书等。审核通过后,CAB将颁发证书。此后,器械注册的最终文档,包括通用提交档案模板 (CSDT)、CAB 证书和申请,以电子方式 [SP1] 在线提交给 MDA 以供审查和最终批准。
CAB 和器械注册证书均应每 5 年更新一次。
注意事项
1. 目前马来西亚注册已无需自由销售证书FSC
2. 必须在在马来西亚指定当地代表提交注册
3. 1个产品只能有1个产品许可证持有人(即持证人)
4. 产品许可证可以转移给其他持有人
5.质量管理体系的要求是什么?
所有制造商都需要 ISO 13485
授权代表AR、进口商、分销商需要良好的分销实践医疗器械 (GDPMD) 认证。
6. 一个产品可以有一个以上进口商或分销商
4 审核流程