FDA 对 I 类和未分类设备的 UDI 要求
2022-07-26
FDA 独特的设备识别系统(UDI 系统)旨在通过分销和使用充分识别设备。其要求旨在根据主要基于设备分类的既定合规日期在七年内分阶段实施。
2013 年 9 月 24 日,FDA 发布了建立 UDI 系统的最终规则,该系统旨在通过分销和使用充分识别设备(“UDI 规则”)。
2018 年 1 月,FDA 发布了本指导文件的初始版本,“唯一设备标识:关于 I 类和非分类设备合规日期的政策”,随后于 2020 年 7 月被同名指南(以下简称“2020 UDI 合规政策指南”)取代。
2022年7月25日,FDA再次更新该指南文件《UDI:关于 I 类和未分类设备的合规日期、直接标记和某些设备的 GUDID 要求的政策》
本指南反映了 FDA 的信念,即继续集中 FDA 的资源解决 UDI 实施问题和高风险设备的数据质量非常重要。
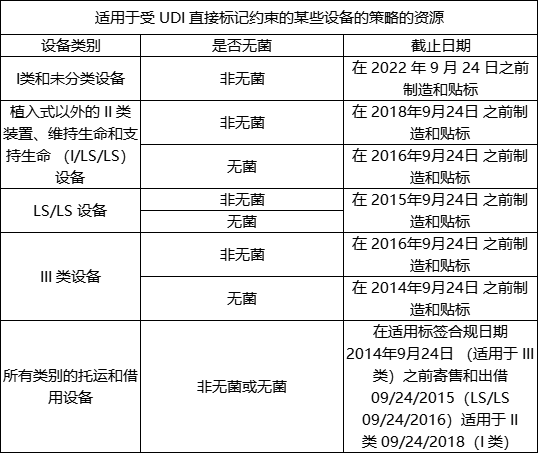
FDA 当时对 UDI 时间的安排:
目前,FDA 不打算根据 21 CFR 830.300 对被视为消费者健康产品的 I 类设备强制执行 GUDID 提交要求,这些设备需要在其标签和设备包装上带有 UDI。就本指南而言,“消费者保健产品”是指 510(k) 豁免的 I 类设备,直接在实体店和/或在线商店的柜台销售给消费者。
FDA 已确定我们不认为是消费者保健产品的 I 类设备可能对公众健康构成更大风险,根据 FDA 的分析,GUDID 数据对于监控这些设备的安全性尤为重要。这些潜在的高风险设备通常专门用于专业医疗机构,并且通常受到额外的监管控制。
FDA 不打算在 2022 年 12 月 8 日之前对 I 类和未分类器械执行 GUDID 提交要 LS/LS 植入式/生命支持或生命维持器械除外。相较于 2020 年指南增加了 75 天。
从 FDA 针对 UDI 指南频繁更新的情况来看,FDA 对于推进 UDI 的重视,我们建议国内各大制造商应尽早完成 UDI 的建立,避免影响正常销售。
